UShER allows researchers to quickly see how a new viral sequence is related to all other variants of SARS-CoV-2, crucial information for tracking transmission dynamics
Tim Stephens | May 10, 2021 | UCSC
The COVID-19 pandemic has spurred genomic surveillance of viruses on an unprecedented scale, as scientists around the world use genome sequencing to track the spread of new variants of the SARS-CoV-2 virus. The rapid accumulation of viral genome sequences presents new opportunities for tracing global and local transmission dynamics, but analyzing so much genomic data is challenging.
“There are now more than a million genome sequences for SARS-CoV-2. No one had anticipated that number when we started sequencing this virus,” said Russ Corbett-Detig, assistant professor of biomolecular engineering at UC Santa Cruz.
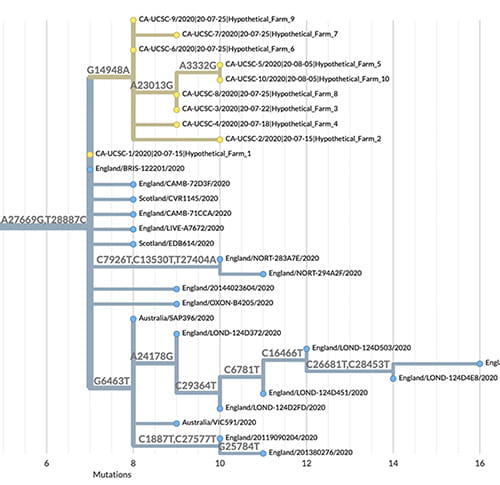
The sheer number of coronavirus genome sequences and their rapid accumulation makes it hard to place new sequences on a “family tree” showing how they are all related. But Corbett-Detig’s group at the UC Santa Cruz Genomics Institute has developed a new method that does this with unprecedented speed. Called Ultrafast Sample Placement on Existing Trees (UShER), this powerful tool is described in a paper published May 10 in Nature Genetics.
UShER identifies the relationships between a user’s newly sequenced viral genomes and all known SARS-CoV-2 virus genomes by adding them to an existing phylogenetic tree, a branching diagram like a family tree that shows how the virus has evolved in different lineages as it accumulates mutations.
“We are able to maintain a comprehensive phylogenetic tree of more than 1.2 million coronavirus sequences and update it with new sequences in real time. No other tool can handle trees of this size with a comparable efficiency,” said first author Yatish Turakhia, a postdoctoral scholar at the Genomics Institute. “This helps us keep track of all variants in circulation, including new variants that are emerging.”