Rachael E. Workman, Alison D. Tang, Paul S. Tang, Miten Jain, John R. Tyson, Roham Razaghi, Philip C. Zuzarte, Timothy Gilpatrick, Alexander Payne, Joshua Quick, Norah Sadowski, Nadine Holmes, Jaqueline Goes de Jesus, Karen L. Jones, Cameron M. Soulette, Terrance P. Snutch, Nicholas Loman, Benedict Paten, Matthew Loose, Jared T. Simpson, Hugh E. Olsen, Angela N. Brooks, Mark Akeson & Winston Timp
Abstract
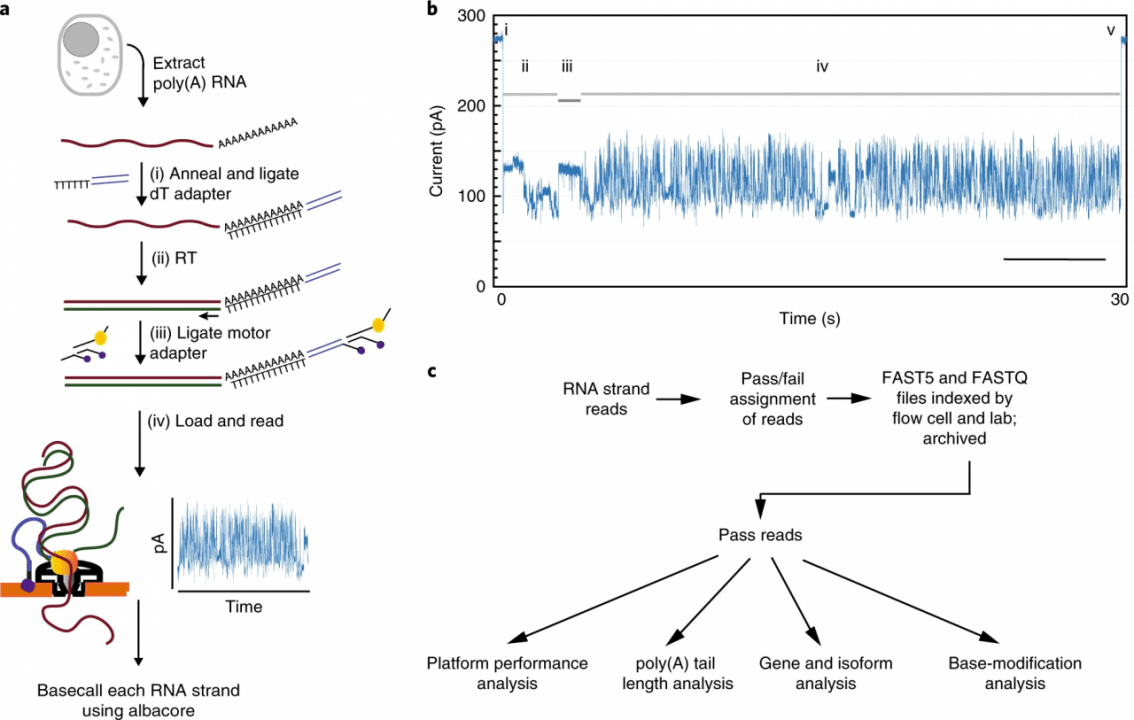
High-throughput complementary DNA sequencing technologies have advanced our understanding of transcriptome complexity and regulation. However, these methods lose information contained in biological RNA because the copied reads are often short and modifications are not retained. We address these limitations using a native poly(A) RNA sequencing strategy developed by Oxford Nanopore Technologies. Our study generated 9.9 million aligned sequence reads for the human cell line GM12878, using thirty MinION flow cells at six institutions. These native RNA reads had a median length of 771 bases, and a maximum aligned length of over 21,000 bases. Mitochondrial poly(A) reads provided an internal measure of read-length quality. We combined these long nanopore reads with higher accuracy short-reads and annotated GM12878 promoter regions to identify 33,984 plausible RNA isoforms. We describe strategies for assessing 3′ poly(A) tail length, base modifications and transcript haplotypes.