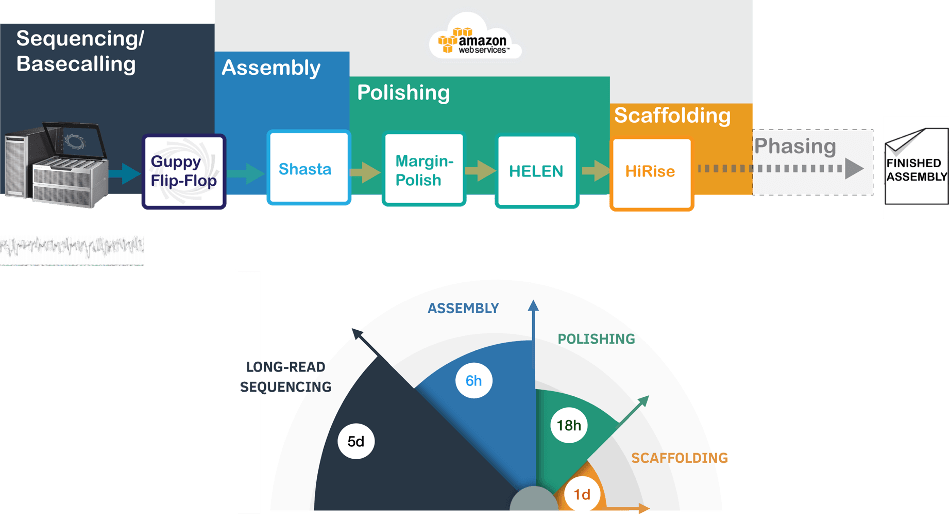
De novo assembly of human genomes using long reads has significant resource overhead. In our recent work, we demonstrate nanopore sequencing and a novel de novo assembly tool Shasta-MarginPolish-HELEN to achieve the de novo assembly of eleven human genomes in nine-days
Kishwar Shafin | University of California, Santa Cruz | May 4, 2020
When choosing a strategy for genome sequencing, scientists face two common choices: “Reference-based analysis or de novo assembly?” and “Long-reads or short-reads?”
Unsurprisingly, most choose reference-based methods using short-reads because they are fast, cheap, easy and proven.
However, the devil is definitely in the details. Short-reads alone fail to generate contiguous assemblies of large genomes because the reads are simply too short1. Using existing scaffolds of an existing reference, short-read can accurately find most small variations2. Although finding small variations made short-reads the genomic workhorse, variant-identification only works in the unique portions of the reference genome, leaving out duplicated and repetitive sequences2. Also, reference-based methods naturally detect alleles similar to the reference, introducing reference allele bias3. This bias is strong for structural variations, which are often missed or miscalled. Short reads often can’t span neighboring variations, so while genotypes are ascertained, the phasing relationships (allelic organization along the maternal and paternal chromosomes) are not4. Until recently long-read de novo assembly methods were expensive, time-consuming, and generally reserved for new species. We asked ourselves: can these challenges be overcome using long-reads and cheap, fast, and scalable methods?
In 2014, the phone-sized, low-cost, Oxford Nanopore MinION was released which democratized third-generation sequencing by making it accessible. In 2017, we participated in an international effort that produced the first reported de novo assembly of a human genome (HG001) using nanopore sequencing. This effort used 53 MinION flowcells, 150,000 CPU hours, and weeks of wall-clock time. While promising, this long-read assembly approach had extensive computational and sequencing requirements5.
In 2018, the high-throughput Oxford Nanopore PromethION device was released. To assess its performance, we sequenced eleven human genomes in nine days and achieved 60x coverage (with ~7x coverage in 100kb+ reads) per sample. This unprecedented sequencing speed required improvements in genome assembly methods. Contemporary approaches for nanopore data-based assemblies required one week of wall-clock time and cost ~$1000.