David Porubsky, Peter Ebert, Peter A. Audano, Mitchell R. Vollger, William T. Harvey, Katherine M. Munson, Melanie Sorensen, Arvis Sulovari, Marina Haukness, Maryam Ghareghani, Human Genome Structural Variation Consortium, Peter M. Lansdorp, Benedict Paten, Scott E. Devine, Ashley D. Sanders, Charles Lee, Mark J.P. Chaisson, Jan O. Korbel, Evan E. Eichler, Tobias Marschall
Abstract
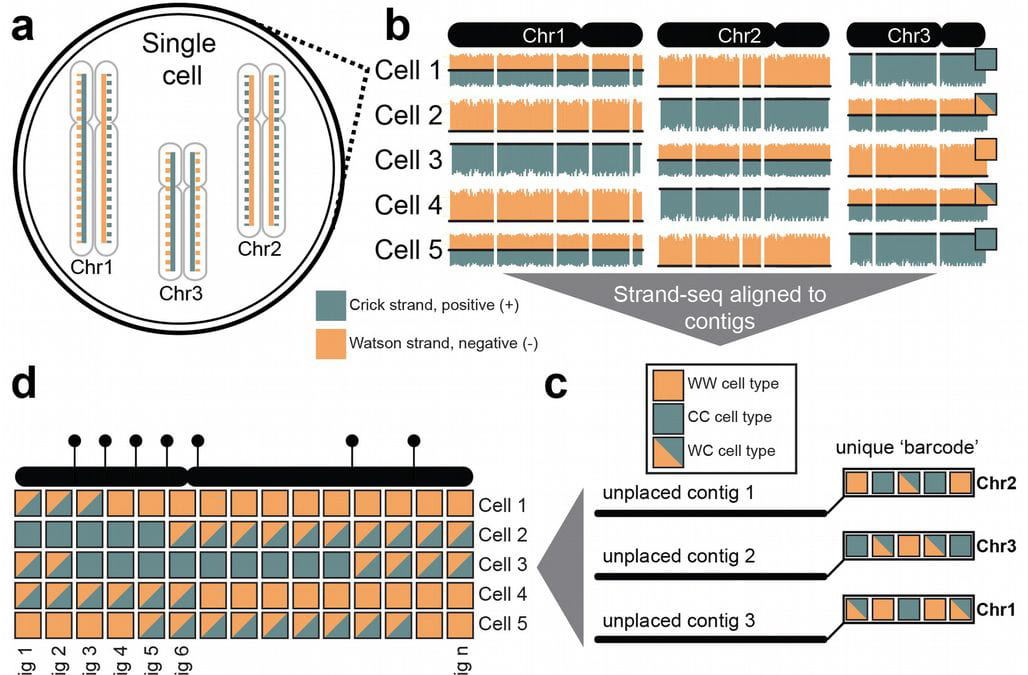
The prevailing genome assembly paradigm is to produce consensus sequences that “collapse” parental haplotypes into a consensus sequence. Here, we leverage the chromosome-wide phasing and scaffolding capabilities of single-cell strand sequencing (Strand-seq)1,2 and combine them with high-fidelity (HiFi) long sequencing reads3, in a novel reference-free workflow for diploid de novo genome assembly. Employing this strategy, we produce completely phased de novo genome assemblies separately for each haplotype of a single individual of Puerto Rican origin (HG00733) in the absence of parental data. The assemblies are accurate (QV > 40), highly contiguous (contig N50 > 25 Mbp) with low switch error rates (0.4%) providing fully phased single-nucleotide variants (SNVs), indels, and structural variants (SVs). A comparison of Oxford Nanopore and PacBio phased assemblies identifies 150 regions that are preferential sites of contig breaks irrespective of sequencing technology or phasing algorithms.